Home_ru
Добро пожаловать в документацию MetaFX!
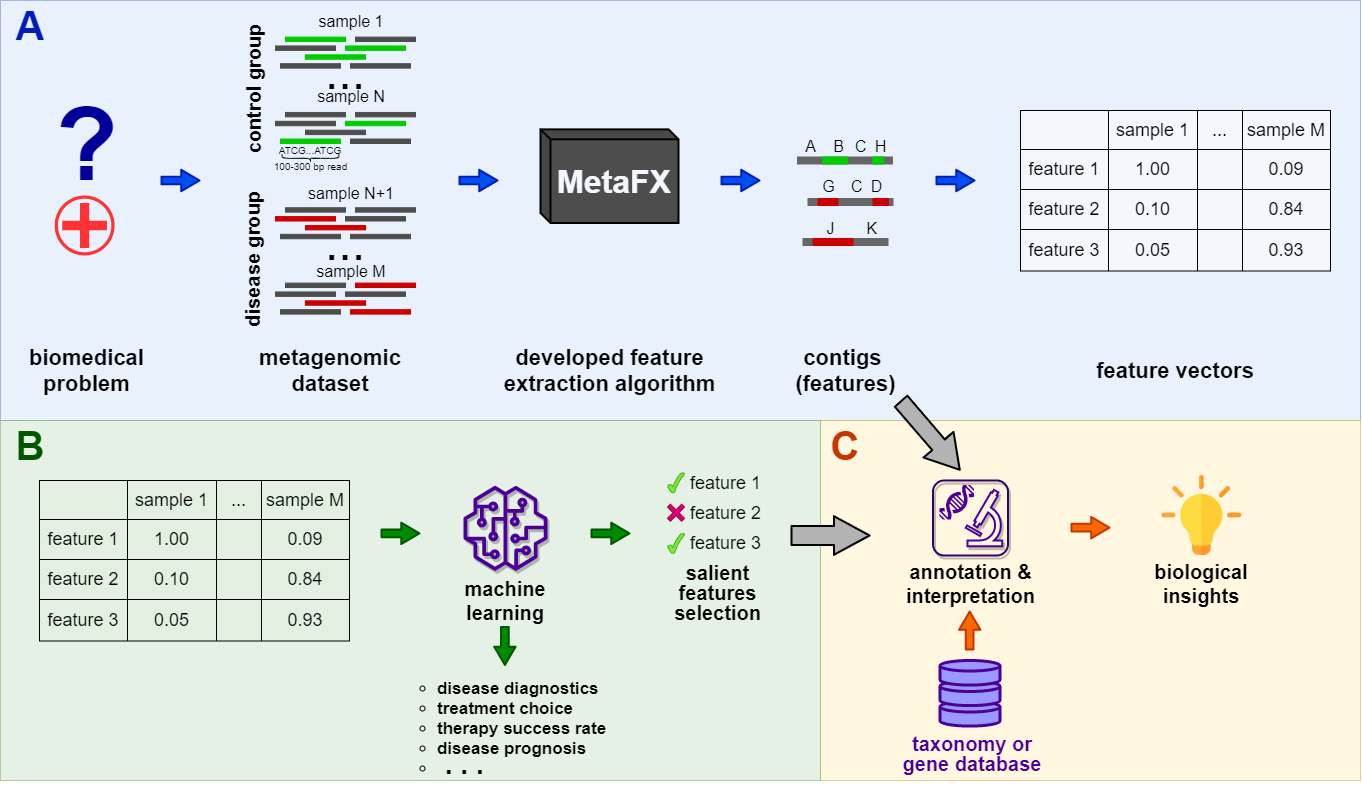
MetaFX (METAgenomic Feature eXtraction) – открытая библиотека для извлечения признаков из метагеномных данных полногеномного секвенирования и классификации групп образцов.
MetaFX основан на идее разработки алгоритма извлечения признаков специфичного для метагеномных данных в виде коротких прочтений. Он позволяет обрабатывать сотни образцов одновременно, каждый размером 1-10 Гб. Отличительной особенностью алгоритма является построение интерпретируемых признаков, которые могут быть использованы не только для тренировки классификационной модели, но и для биологической аннотации и интрепретации.
Для запуска MetaFX, необходимо скачать репозиторий проекта с запускаемыми файлами.
git clone https://github.com/ctlab/metafx
cd metafxИ добавить директорию в системную переменную PATH.
export PATH=/path/to/metafx/bin:/path/to/metafx/bin/metafx-modules:/path/to/metafx/bin/metafx-scripts:$PATHДля постоянного использования будет удобно добавить эту команду в файл ~/.profile или ~/.bashrc.
Зависимости:
- JRE версии 1.8 или выше
- python=3.9.5 (может быть удобно создать отдельное окружение conda для установки всех зависимостей)
conda create -n metafx_env python=3.9.5- библиотеки python указаны в файле
requirements.txtfile. Они могут быть установлены с использованием pip
python -m pip install --upgrade pip
pip install -r requirements.txt-
coreutils требуется для платформы macOS (e.g.
brew install coreutils) - Для запуска модуля
metafx metaspades, необходима программа SPAdes. Для установки следуйте инструкции (не рекомендуется для первого раза). - Для запуска модуля
metafx bandage, неоходима программа BandageNG. Для установки следуйте инструкции.
Код был протестирован для операционных систем Ubuntu 18.04 LTS, Ubuntu 20.04 LTS, macOS 11 Big Sur, and macOS 12 Monterey, и должен выполняться на системах Linux/macOS.
Для многопоточного запуска и ускорения работы необходимо минимум два вычислительных потока. Объем оперативной памяти линейно зависит от размера обрабатываемых файлов со входными прочтениями. Дополнительное место на жестком диске для промежуточных файлов также линейно зависит от объема входных данных. Например, для туториала с объемом входных файлов 12 Гб требуется 16 Гб памяти на жестком диске для промежуточных вычислений и 8 Гб оперативной памяти. При использовании 6 потоков, время исполнения туториала приблизительно равняется одному часу.
Для запуска MetaFX используйте следующий синтаксис:
metafx <pipeline> [<Launch options>] [<Input parameters>]Для просмотра всех поддерживаемых модулей запустите metafx -h или metafx --help.
Для просмотра опций и входных параметров конкретного модуля запустите metafx <pipeline> -h или metafx <pipeline> --help.
MetaFX поддерживает как парные так и одноконцевые прочтения. Для корректного автоматического распознавания парных прочтений, файлы должны именоваться с суффиксами "_R1"&"_R2" или "_r1"&"_r2" в конце названия перед расширением. Например, sample_r1.fastq&sample_r2.fastq, или reads_R1.fq.gz&reads_R2.fq.gz.
При запуске MetaFX создается рабочая директория (по умолчанию ./workDir/). В ней сохраняются все результаты и промежуточные файлы.
MetaFX содержит множество модулей, которые можно разбить на три группы:
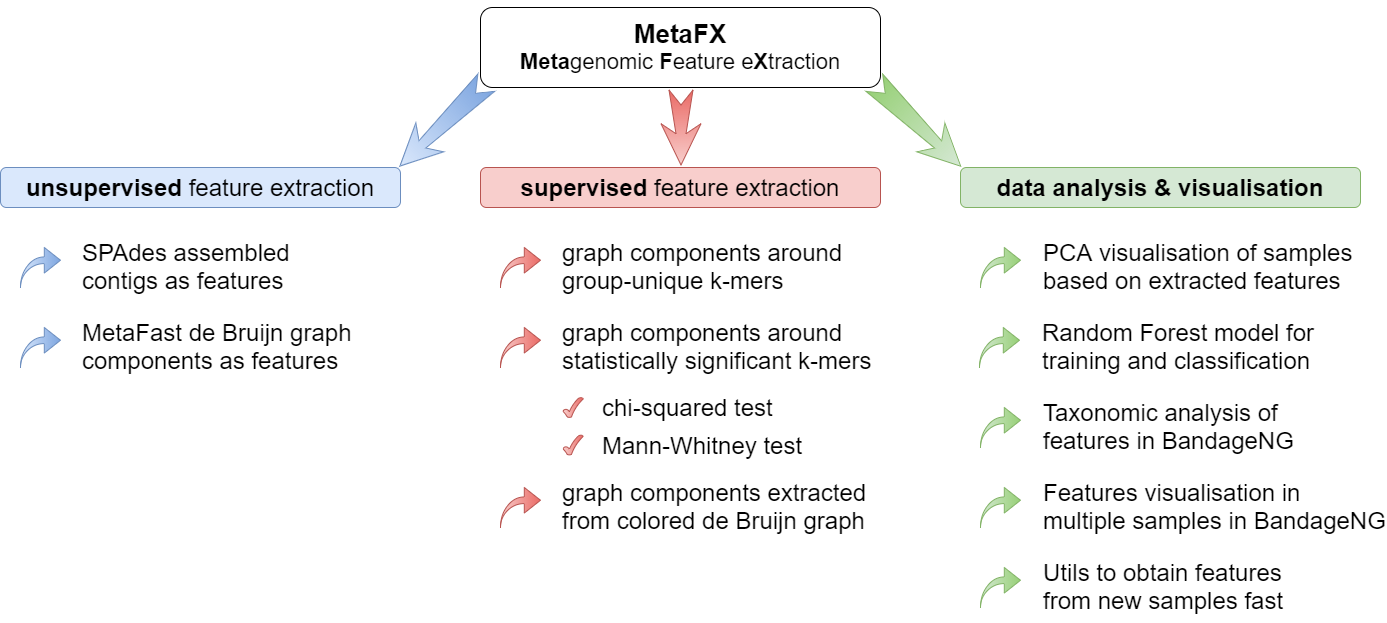
Алгоритмы направлены на извлечение признаков из метагеномного набора данных без использования вспомогательной информации об образцах. Алгоритмы производят (псевдо-)сборку образцов по-отдельности и строят общий граф де Брейна. Далее граф разбивается на компоненты, которые используются в качестве признаков.
Производит извлечения признаков «без учителя» и оценку близости образцов на основе алгоритма MetaFast.
metafx metafast -t 2 -m 4G -w wd_metafast -k 21 -i test_data/test*.fastq.gz -b1 100 -b2 500Входные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filenames> | Список файлов с прочтениями. FASTQ, FASTA, gzip- or bzip2-compressed [обязательно] |
| -b, --bad-frequency <int> | максимальная частота встречаемости ошибочных k-меров [по умолчанию: 1] |
| -l, --min-seq-len <int> | минимальная длина последовательности в компоненте (в нуклеотидах) [по умолчанию: 100] |
| -b1, --min-comp-size <int> | минимальный размер компоненты (признака) в k-мерах [по умолчанию: 1000] |
| -b2, --max-comp-size <int> | максимальный размер компоненты (признака) в k-мерах [по умолчанию: 10000] |
| --kmers-dir <dirname> | папка с предподсчитанными k-мерами для образцов в двоичном формате [необязательно] |
| --skip-graph | если выставлен, пропустить шаг построения графа де Брейна и fasta файлов для извлеченных компонент [по умолчанию: False] |
Выходные файлы
Для обратной совместимости с методами извлечения признаков «с учителем», каждый образце записывется в категорию all.
| файл | описание |
|---|---|
| wd_metafast/categories_samples.tsv | файл с 3 столбцами: <category>\t<present_samples>\t<absent_samples> |
| wd_metafast/samples_categories.tsv | файл с 2 столбцами: <sample_name>\t<category> |
| wd_metafast/feature_table.tsv | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы |
| wd_metafast/contigs_all/components.seq.fasta | контиги-признаки в формате FASTA (для аннотации и интрепретации) |
| wd_metafast/contigs_all/components-graph.gfa | де Брюйн граф признаков в формате GFA (для визуализации в BandageNG) |
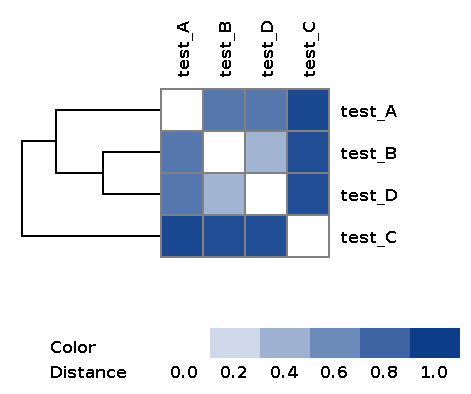
| wd_metafast/matrices/dist_matrix_<DATE_TIME>.txt | Матрица расстояний между каждой парой образцов на основе признаков по несходству Брэя-Кертиса |
| wd_metafast/matrices/dist_matrix_<DATE_TIME>_heatmap.png | Тепловая карта и дендрограмма близости образцов построенные по матрице расстояний |
Пример полученной дендрограммы:
Производит извлечения признаков «без учителя» и оценку близости образцов на основе алгоритма metaSpades
NB: Требуется установка SPAdes. Пожалуйста, следуйте инструкции
metafx metaspades -t 2 -m 4G -w wd_metaspades -k 21 -i test_data/test_*.fastq.gz -b1 500 -b2 5000Входные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filenames> | список файлов с парными прочтениями. FASTQ, FASTA, gzip-compressed [обязательно] |
| --separate | если выставлен, использовать каждый контиг в качестве отдельного признака (-l, -b1, -b2 будут проигнорированы) [по умолчанию: False] |
| -l, --min-seq-len <int> | минимальная длина последовательности в компоненте (в нуклеотидах) [по умолчанию: 100] |
| -b1, --min-comp-size <int> | минимальный размер компоненты (признака) в k-мерах [по умолчанию: 1000] |
| -b2, --max-comp-size <int> | максимальный размер компоненты (признака) в k-мерах [по умолчанию: 10000] |
| --kmers-dir <dirname> | папка с предподсчитанными k-мерами для образцов в двоичном формате [необязательно] |
| --skip-graph | если выставлен, пропустить шаг построения графа де Брейна и fasta файлов для извлеченных компонент [по умолчанию: False] |
Выходные файлы
Для обратной совместимости с методами извлечения признаков «с учителем», каждый образце записывется в категорию all.
| файл | описание |
|---|---|
| wd_metaspades/categories_samples.tsv | файл с 3 столбцами: <category>\t<present_samples>\t<absent_samples> |
| wd_metaspades/samples_categories.tsv | файл с 2 столбцами: <sample_name>\t<category> |
| wd_metaspades/feature_table.tsv | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы |
| wd_metaspades/contigs_all/components.seq.fasta | контиги-признаки в формате FASTA (для аннотации и интрепретации) |
| wd_metaspades/contigs_all/components-graph.gfa | де Брюйн граф признаков в формате GFA (для визуализации в BandageNG) |
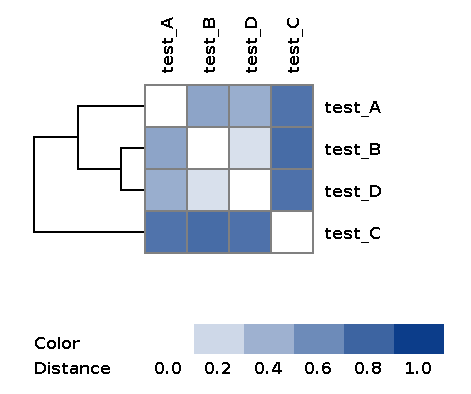
| wd_metaspades/matrices/dist_matrix_<DATE_TIME>.txt | Матрица расстояний между каждой парой образцов на основе признаков по несходству Брэя-Кертиса |
| wd_metaspades/matrices/dist_matrix_<DATE_TIME>_heatmap.png | Тепловая карта и дендрограмма близости образцов построенные по матрице расстояний |
Пример полученной дендрограммы:
Алгоритмы направлены на извлечение признаков из метагеномного набора данных специфичных для групп образцов с использованием вспомогательной информации об образцах, такой как диагноз, терапия, результаты анализов и другой. Набор образцов разбивается на группы на основе метаданных и для каждой группы извлекаются специфичные признаки на основе графа де Брейна. Затем все признаки объединяются в одну таблицу.
Производит извлечение признаков на основе специфичных k-меров. K-мер является специфичным для группы при условии присутствия хотя бы в G образцах указанной группы и отсутствии во всех образцах остальных групп.
metafx unique -t 2 -m 4G -w wd_unique -k 21 -i test_data/sample_list_train.txtВходные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filename> | файл с двумя значениями в каждой строке: <path_to_file>\t<category> [обязательно] |
| -b, --bad-frequency <int> | максимальная частота встречаемости ошибочных k-меров [по умолчанию: 1] |
| --min-samples <int> | k-мер специфичен если встречается хотя бы в G образцах. G изменяется в диапазоне [--min-samples; --max-samples] [по умолчанию: 2] |
| --max-samples <int> | k-мер специфичен если встречается хотя бы в G образцах. G изменяется в диапазоне [--min-samples; --max-samples] [по умолчанию: #{samples in category}/2 + 1] |
| --depth <int> | глубина обхода графа де Брейна по развилкам от опрного k-мера[по умолчанию: 1] |
| --kmers-dir <dirname> | папка с предподсчитанными k-мерами для образцов в двоичном формате [необязательно] |
| --skip-graph | если выставлен, пропустить шаг построения графа де Брейна и fasta файлов для извлеченных компонент [по умолчанию: False] |
Выходные файлы
| файл | описание |
|---|---|
| wd_unique/categories_samples.tsv | файл с 3 столбцами: <category>\t<present_samples>\t<absent_samples> |
| wd_unique/samples_categories.tsv | файл с 2 столбцами: <sample_name>\t<category> |
| wd_unique/feature_table.tsv | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы |
| wd_unique/contigs_<category>/components.seq.fasta | контиги-признаки в формате FASTA (для аннотации и интрепретации) |
| wd_unique/contigs_<category>/components-graph.gfa | де Брюйн граф признаков в формате GFA (для визуализации в BandageNG) |
Производит извлечение признаков на основе теста хи-квадрат и отбирает заданное число наиболее значимых k-меров. Значимость k-мера для группы определяется в два этапа. На первом этапе все k-меры сортируются по значимости на основе значения p-value полученного в результате теста хи-квадрат. На втором этапе отбирается наиболее значимые N k-меров для каждой группы (всего N в случае двух и трех групп).
metafx chisq -t 2 -m 4G -w wd_chisq -k 21 -i test_data/sample_list.txt -n 1000Входные параметры
| параметр | description |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filename> | файл с двумя значениями в каждой строке: <path_to_file>\t<category> [обязательно] |
| -n, --num-kmers <int> | число извлекаемых специфичных k-меров [обязательно] |
| -b, --bad-frequency <int> | максимальная частота встречаемости ошибочных k-меров [по умолчанию: 1] |
| --depth <int> | глубина обхода графа де Брейна по развилкам от опрного k-мера[по умолчанию: 1] |
| --kmers-dir <dirname> | папка с предподсчитанными k-мерами для образцов в двоичном формате [необязательно] |
| --skip-graph | если выставлен, пропустить шаг построения графа де Брейна и fasta файлов для извлеченных компонент [по умолчанию: False] |
Выходные файлы
| файл | описание |
|---|---|
| wd_chisq/categories_samples.tsv | файл с 3 столбцами: <category>\t<present_samples>\t<absent_samples> |
| wd_chisq/samples_categories.tsv | файл с 2 столбцами: <sample_name>\t<category> |
| wd_chisq/feature_table.tsv | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы |
| wd_chisq/contigs_<category>/components.seq.fasta | контиги-признаки в формате FASTA (для аннотации и интрепретации) |
| wd_chisq/contigs_<category>/components-graph.gfa | де Брюйн граф признаков в формате GFA (для визуализации в BandageNG) |
Производит извлечение признаков на основе статистически значимых k-меров. Значимость k-мера определяется в два этапа. На первом этапе с помощью теста Пирсона хи-квадрат на равномерность отфильтровываются k-меры, распределение которых совпадает между образцами из разных групп. На втором этапе применяется U тест Манна-Уитни чтобы отобрать k-меры с разной частотой встречаемости между категориями.
metafx stats -t 2 -m 4G -w wd_stats -k 21 -i test_data/sample_list.txt --pchi2 0.05 --pmw 0.05Входные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filename> | файл с двумя значениями в каждой строке: <path_to_file>\t<category> [обязательно] |
| -b, --bad-frequency <int> | максимальная частота встречаемости ошибочных k-меров [по умолчанию: 1] |
| --pchi2 <float> | p-value для теста хи-квадрат [по умолчанию: 0.05] |
| --pmw <float> | p-value для теста Манна-Уитни [по умолчанию: 0.05] |
| --depth <int> | глубина обхода графа де Брейна по развилкам от опрного k-мера[по умолчанию: 1] |
| --kmers-dir <dirname> | папка с предподсчитанными k-мерами для образцов в двоичном формате [необязательно] |
| --skip-graph | если выставлен, пропустить шаг построения графа де Брейна и fasta файлов для извлеченных компонент [по умолчанию: False] |
Выходные файлы
| файл | описание |
|---|---|
| wd_stats/categories_samples.tsv | файл с 3 столбцами: <category>\t<present_samples>\t<absent_samples> |
| wd_stats/samples_categories.tsv | файл с 2 столбцами: <sample_name>\t<category> |
| wd_stats/feature_table.tsv | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы |
| wd_stats/contigs_<category>/components.seq.fasta | контиги-признаки в формате FASTA (для аннотации и интрепретации) |
| wd_stats/contigs_<category>/components-graph.gfa | де Брюйн граф признаков в формате GFA (для визуализации в BandageNG) |
Производит извлечение признаков на основе раскрашенного графа де Брейна. Все k-меры раскрашиваются на основании относительной частоты встречаемости в образцах разных групп. Затем граф де Брейна разбивается на раскрашенные компоненты.
Важно! Данный модуль поддерживает до трех групп образцов.
metafx colored -t 2 -m 4G -w wd_colored -k 21 -i test_data/sample_list_3.txt --linear --n-comps 100 --perc 0.8Входные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filename> | файл с двумя значениями в каждой строке: <path_to_file>\t<category> [обязательно] |
| -b, --bad-frequency <int> | максимальная частота встречаемости ошибочных k-меров [по умолчанию: 1] |
| --total-coverage | если выставлен, частота встречаемости k-меров рассчитывается как сумма покрытия в образцах, иначе как число образцов [по умолчанию: False] |
| --separate | если выставлен, использовать только раскрашенные k-меры в компонентах (на работает в случае --linear) [по умолчанию: False] |
| --linear | если выставлен, извлекать только линейные пути в качестве компонент [по умолчанию: False] |
| --n-comps <int> | выбирать не больше чем <int> компонент для каждой категории [по умолчанию: -1, означает все компоненты] |
| --perc <float> | частота встречаемости k-мера для определения его специфичности [по умолчанию: 0.9] |
| --kmers-dir <dirname> | папка с предподсчитанными k-мерами для образцов в двоичном формате [необязательно] |
| --skip-graph | если выставлен, пропустить шаг построения графа де Брейна и fasta файлов для извлеченных компонент [по умолчанию: False] |
Выходные файлы
| файл | описание |
|---|---|
| wd_colored/categories_samples.tsv | файл с 3 столбцами: <category>\t<present_samples>\t<absent_samples> |
| wd_colored/samples_categories.tsv | файл с 2 столбцами: <sample_name>\t<category> |
| wd_colored/feature_table.tsv | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы |
| wd_colored/contigs_<category>/components.seq.fasta | контиги-признаки в формате FASTA (для аннотации и интрепретации) |
| wd_colored/contigs_<category>/components-graph.gfa | де Брюйн граф признаков в формате GFA (для визуализации в BandageNG) |
Алгоритмы направлены на анализ результатов извлечения признаков. Есть методы для визуализации близости образцов и обучения классификационных моделей. Модели используют извлеченные признаки для предсказания свойств образцов и классификации новых образцов.
Результаты одного из методов извлечения признаков MetaFX требуются для дальнейшего анализа.
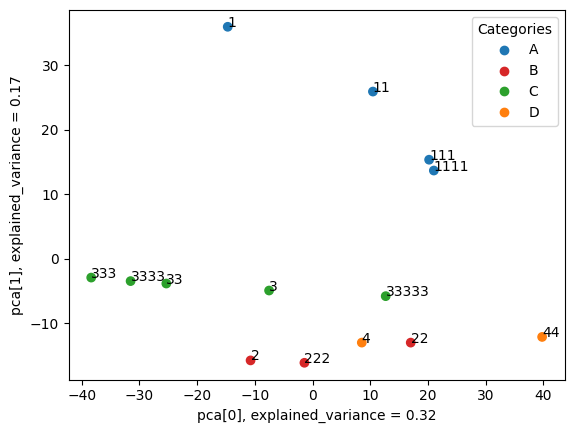
Производит уменьшение размерности PCA и визуализацию близости образцов на основе извлеченных признаков.
metafx pca -w wd_pca -f wd_stats/feature_table.tsv -i wd_stats/samples_categories.tsv --showВходные параметры
| параметр | описание |
|---|---|
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -f, --feature-table <filename> | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы (используйте "workDir/feature_table.tsv") [обязательно] |
| -i, --metadata-file <filename> | файл с 2 столбцами: <sample_name>\t<category> (используйте "workDir/samples_categories.tsv") [необязательно, по умолчанию: None] |
| --name <filename> | имя итогового изображения [необязательно, по умолчанию: pca] |
| --show | если выставлен, отображать названия образцов на графике [необязательно, по умолчанию: False] |
Выходные файлы
| файл | описание |
|---|---|
| wd_pca/pca.png | график 2-мерного PCA в формате png |
| wd_pca/pca.svg | график 2-мерного PCA в формате svg |
Пример графика PCA:
Обучение классификатора с использованием машинного обучения на основе извлеченных признаков.
metafx fit -w wd_fit -f wd_unique/feature_table.tsv -i wd_unique/samples_categories.tsvВходные параметры
| параметр | описание |
|---|---|
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -f, --feature-table <filename> | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы (используйте "workDir/feature_table.tsv") [обязательно] |
| -e, --estimator [RF, XGB, Torch] | классификатор: RF – scikit-learn Random Forest, XGB – XGBoost, Torch – PyTorch neural network, [необязательно, по умолчанию: RF] |
| -i, --metadata-file <filename> | файл с 2 столбцами: <sample_name>\t<category> (используйте "workDir/samples_categories.tsv") [необязательно, по умолчанию: None] |
| --name <filename> | имя итоговой модели [необязательно, по умолчанию: rf_model] |
Выходные файлы
| файл | описание |
|---|---|
| wd_fit/rf_model.joblib | бинарный файл с предобученной классификационной моделью для использования в модуле metafx predict
|
Используется для обработки новых признаков и расчета численных значений извлеченных признаков. Используется в случае обработки новых данных. Вначале из набора данных с известными метками извлекаются признаки и обучается модель. Затем для новых образцов формируется таблица признаков с помощью метода calc_features и получаются предсказания с помощью метода predict.
metafx calc_features -t 2 -m 4G -w wd_calc_features -k 21 -d wd_unique \
-i test_data/test_A_R1.fastq.gz test_data/test_A_R2.fastq.gz \
test_data/test_B_R1.fastq.gz test_data/test_B_R2.fastq.gz \
test_data/test_C_R1.fastq.gz test_data/test_C_R2.fastq.gz Входные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filenames> | Список файлов с прочтениями. FASTQ, FASTA, gzip- or bzip2-compressed [обязательно] |
| -d, --feature-dir <dirname> | папка с файлами components.bin для каждой группы образцов и файлом categories_samples.tsv. Как правило, это результат работы модулей MetaFX (unique, chisq, stats, colored, metafast, metaspades) [обязательно] |
| -b, --bad-frequency <int> | максимальная частота встречаемости ошибочных k-меров [по умолчанию: 1] |
| --kmers-dir <dirname> | папка с предподсчитанными k-мерами для образцов в двоичном формате [необязательно] |
Выходные файлы
| файл | описание |
|---|---|
| wd_calc_features/feature_table.tsv | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы |
Используется классификатор для предсказания категорий новых образцов по предобученной модели.
Как правило, --feature-table получается с помощью модуля MetaFX calc_features и --model с помощью модуля fit или cv.
metafx predict -w wd_predict -f wd_calc_features/feature_table.tsv --model wd_fit/rf_model.joblibВходные параметры
| параметр | описание |
|---|---|
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -f, --feature-table <filename> | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы (используйте "workDir/feature_table.tsv") [обязательно] |
| --model <filename> | файл с предобученной классификационной моделью, в результате модулей fit или cv (используйте "workDir/rf_model.joblib") [обязательно] |
| -i, --metadata-file <filename> | файл с 2 столбцами: <sample_name>\t<category> (используйте "workDir/samples_categories.tsv") [необязательно, по умолчанию: None] |
| --name <filename> | имя файла с предсказанными категориями [необязательно, по умолчанию: predictions] |
Выходные файлы
| файл | описание |
|---|---|
| wd_predict/predictions.tsv | файл с 2 столбцами: <sample name>\t<predicted category> |
Пример выходного файла:
$ cat wd_predict/predictions.tsv
test_A A
test_C C
test_B BКак и ожидалось, категория каждого файла верно определена.
Обучение классификатора с использованием машинного обучения на основе извлеченных признаков и проверка точности с помощью кросс-валидации. Также может производиться поиск наиболее оптимальных параметров классификационной модели.
metafx cv -t 2 -w wd_cv -f wd_stats/feature_table.tsv -i wd_stats/samples_categories.tsv -n 2 --gridВходные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: 1] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -f, --feature-table <filename> | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы (используйте "workDir/feature_table.tsv") [обязательно] |
| -i, --metadata-file <filename> | файл с 2 столбцами: <sample_name>\t<category> (используйте "workDir/samples_categories.tsv") [необязательно, по умолчанию: None] |
| -n, --n-splits <int> | число разбиение в кросс-валидации, хотя бы 2 [необязательно, по умолчанию: 5] |
| --name <filename> | имя итоговой модели [необязательно, по умолчанию: rf_model] |
| --grid | если выставлен, производится поиск оптимальных гиперпараметров для классификатора [необязательно, по умолчанию: False] |
Выходные файлы
| файл | описание |
|---|---|
| wd_cv/rf_model_cv.joblib | бинарный файл с предобученной классификационной моделью для использования в модуле metafx predict
|
Обучение классификатора с использованием машинного обучения на основе извлеченных признаков и классификация новых образцов. Используется когда весь набор данных доступен сразу, но только его часть имеет метки. --feature-table содержит информацию обо всех образцах, --metadata-file содержит информацию только о части образцов.
Для моделирования ситуации используем wd_colored/feature_table.tsv с 12 образцами.
Создадим специальный файл test_labels.tsv с метками только для 6 образцов.
$ echo -e "1\tA\n11\tA\n2\tB\n22\tB\n3\tC\n33\tC" > test_labels.tsv
$ cat test_labels.tsv
1 A
11 A
2 B
22 B
3 C
33 Cmetafx fit_predict -w wd_fit_predict -f wd_colored/feature_table.tsv -i test_labels.tsvВходные параметры
| параметр | описание |
|---|---|
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -f, --feature-table <filename> | файл-матрицы с численными значениями признаков: строки – признаки, столбцы – образцы (используйте "workDir/feature_table.tsv") [обязательно] |
| -i, --metadata-file <filename> | файл с 2 столбцами: <sample_name>\t<category> (используйте "workDir/samples_categories.tsv") [необязательно, по умолчанию: None] |
| --name <filename> | имя итоговой модели [необязательно, по умолчанию: rf_model] |
Выходные файлы
| файл | описание |
|---|---|
| wd_fit_predict/model.joblib | бинарный файл с предобученной классификационной моделью для использования в модуле metafx predict
|
| wd_fit_predict/model.tsv | файл с 2 столбцами: <sample name>\t<predicted category> |
Пример выходного файла:
$ cat wd_fit_predict/model.tsv
111 A
1111 A
222 B
333 C
3333 C
33333 CКак ожидалось, все образцы верно классифицированы.
Подготовка для визуализации классификационной модели и признаков на графе де Брейнав приложении BandageNG. Может обучать классификатор с нуля.
Требуется установка приложения BandageNG:
- Загрузите бинарный файл (Linux or macOS): https://github.com/ctlab/BandageNG/releases
- Переименуйте в
BandageNGи сделайте исполняемым (chmod u+x BandageNG) - добавьте папку с
BandageNGв переменную PATH
export PATH=/path/to/BandageNG/:$PATHmetafx bandage -w wd_bandage -f wd_unique -n 20Входные параметры
| параметр | описание |
|---|---|
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -f, --feature-dir <dirname> | папка с файлами components.bin для каждой группы образцов и файлом categories_samples.tsv. Как правило, это результат работы модулей MetaFX (unique, chisq, stats, colored, metafast, metaspades) [обязательно] |
| --model <filename> | файл с предобученной классификационной моделью, в результате модулей fit или cv (используйте "workDir/rf_model.joblib") [обязательно] |
| -n, --n-estimators <int> | число базовых классификаторов [необязательно] |
| -d, --max-depth <int> | максимальная шлубина дерева решений [необязательно] |
| -e, --estimator [RF, ADA, GBDT] | классификатор: RF – Random Forest, ADA – AdaBoost, GBDT – Gradient Boosted Decision Trees [необязательно, по умолчанию: RF] |
| --draw-graph | если выставлен, производится построение графа де Брейна [по умолчанию: False] |
| --gui | если выставлен, запускает BandageNG. Не работает без графического интерфейса [по умолчанию: False] |
| --name <filename> | имя выходного файла с деревом в текстовом формате [необязательно, по умолчанию: tree_model] |
Выходные файлы
| файл | описание |
|---|---|
| wd_bandage/tree_model.txt | предобученная классификационная модель в текстовом формате для визуализации в BandageNG |
| wd_bandage/tree_model.joblib | бинарный файл с предобученной моделью |
| wd_bandage/spades_graph/assembly_graph_with_scaffolds.gfa | граф де Брейна на признаках для визуализации в BandageNG |
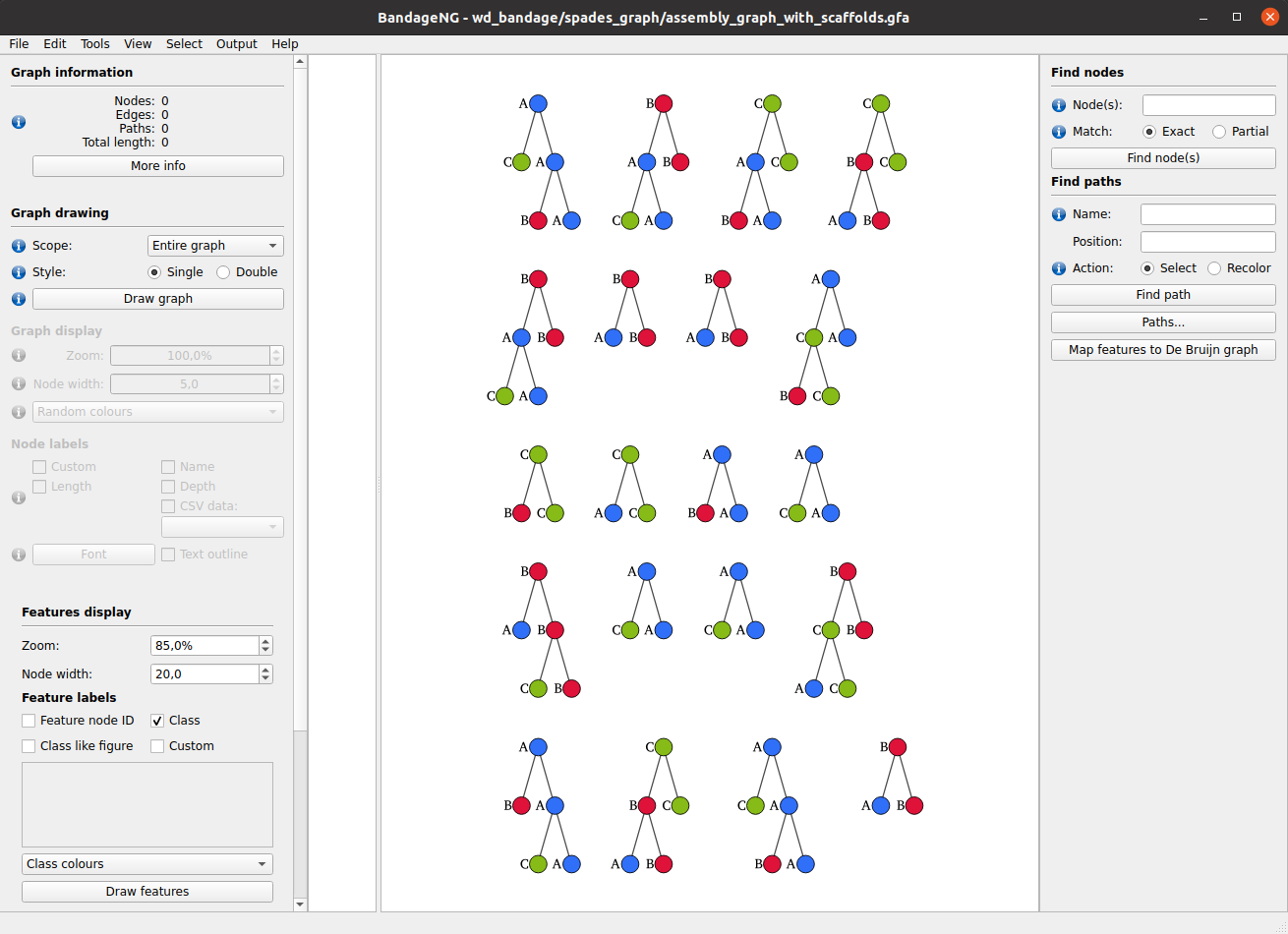
Итоговый файл может быть визуализирован в BandageNG с помощью следующих шагов:
- Откройте BandageNG.
-
File → Load features forest: выберите файл
wd_bandage/tree_model.txt. - На панели слева Features display нажмите кнопку Draw features. Будет отрисована классификационная модель.
-
File → Load graph: выберите файл
wd_bandage/spades_graph/assembly_graph_with_scaffolds.gfa. - На панели слева Graph drawing нажмите кнопку Draw graph. Будет отрисован граф де Брейна.
- Признаки классификационной модели могут быть отмечены на графе. Выберите вершины в деревев правой части экрана и нажмите кнопку Connect feature nodes with de bruijn nodes на панели справа.
В примере мы получаем следующий результат (только первый три шага):
Отбирает образцы с признаком выше заданного порога и строит локальный граф де Брейна для каждого образца с помощью приложения MetaCherchant. Полученные графы могут быть проанализированы в BandageNG в мулитиграфовом режиме.
Все файлы должны быть размещены в одной директории. Например так
mkdir reads ln -s `pwd`/test_data/3* reads/ ln -s `pwd`/test_data/4* reads/ ln -s `pwd`/test_data/test/* reads/
metafx feature_analysis -t 2 -m 4G -w wd_feat_analysis -k 21 -f wd_chisq/ -n A_15 -r reads/ --relab 0.5Входные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -f, --feature-dir <dirname> | папка с контигами для каждой категории, и файлами feature_table.tsv и categories_samples.tsv. Как правило, это результат модулей MetaFX (unique, stats, colored, metafast, metaspades) [обязательно] |
| -n, --feature-name <string> | имя признака (из первого столбца файла feature_table.tsv) [обязательно] |
| -r, --reads-dir <dirname> | папка с файлами прочтений для образцов. FASTQ, FASTA, gzip- or bzip2-compressed [обязательно] |
| --relab <int> | минимальное относительное покрытие признака для включения образца в анализ [необязательно, по умолчанию: 0.1] |
Выходные файлы
| файл | описание |
|---|---|
| wd_feat_analysis/samples_list_feature_<feature-name>.txt | список образцов содержащих признак |
| wd_feat_analysis/seq_feature_<feature-name>.fasta | нуклеотидная последовательность признака в формате FASTA |
| wd_feat_analysis/graphs/ | папка с графами для отобранных образцов |
Все графы сохраняются в папке ${w}/graphs/ и могут быть одновременно визуализированы в BandageNG.
Необходима установка BandageNG:
- Загрузите бинарный файл (Linux or macOS): https://github.com/ctlab/BandageNG/releases
- Переименуйте в
BandageNGи сделайте исполняемым (chmod u+x BandageNG) - добавьте папку с
BandageNGв переменную PATH
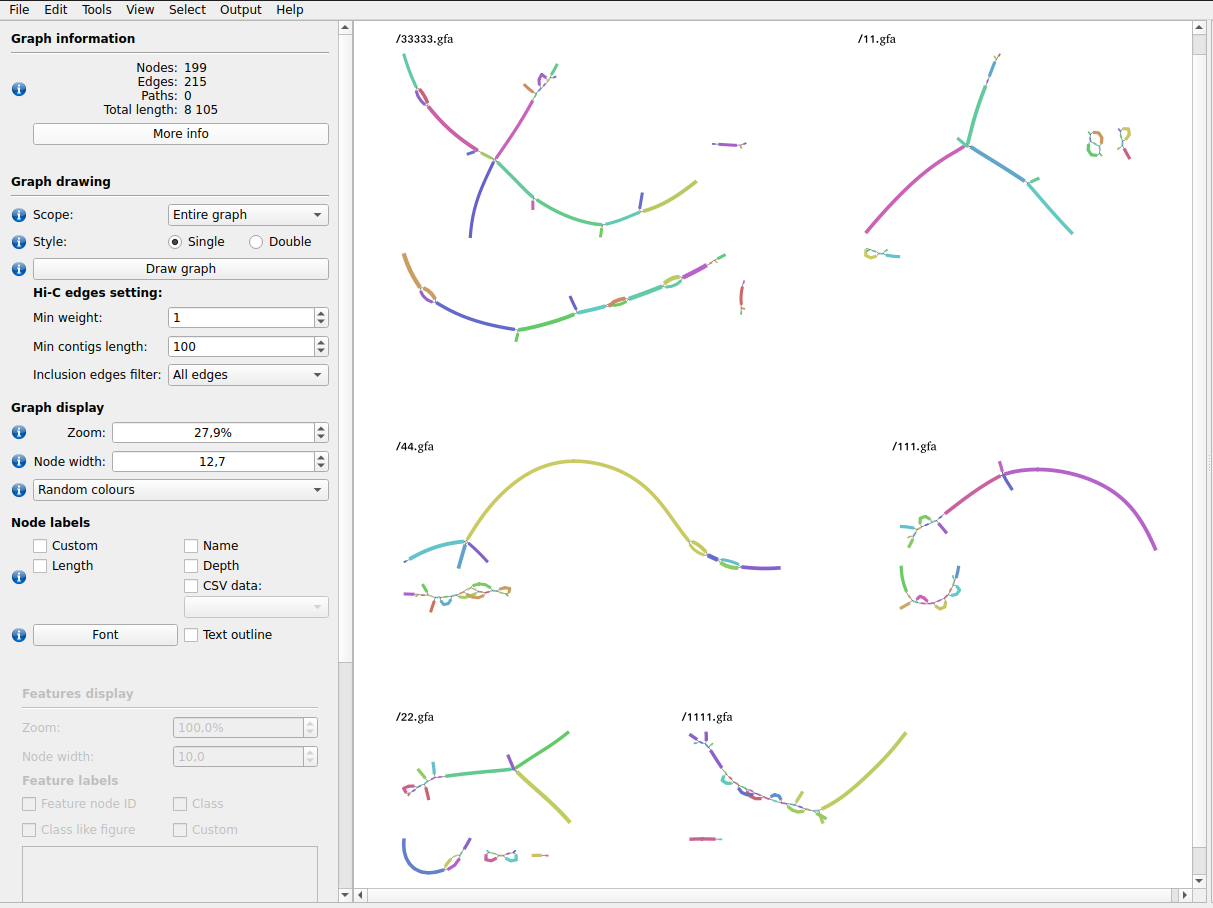
export PATH=/path/to/BandageNG/:$PATHВизуализация полученных графов в BandageNG BandageNG (подробная инструкция):
- Откройте BandageNG.
-
File → Load graphs from dir: выберите папку
wd_feat_analysis/graphs/directory. - На панели слева Graph drawing нажмите кнопку Draw graph. Будут отрисованы все графы.
Пример полученного результата:
Модули для работы с данными и ускорения MetaFX.
Предварительная обработка файлов с прочтениями и извлечение из них k-меров.
Ускоряет работу и уменьшает потребление RAM при передаче в параметр --kmers-dir.
metafx extract_kmers -t 2 -m 4G -w wd_extract_kmers -k 21 \
-i test_data/test_A_R1.fastq.gz test_data/test_A_R2.fastq.gz \
test_data/test_B_R1.fastq.gz test_data/test_B_R2.fastq.gz \
test_data/test_C_R1.fastq.gz test_data/test_C_R2.fastq.gzВходные параметры
| параметр | описание |
|---|---|
| -t, --threads <int> | число потоков [по умолчанию: все] |
| -m, --memory <MEM> | объем оперативной памяти (значения с суффиксами: 1500M, 4G, etc.) [по умолчанию: 90% свободной RAM] |
| -w, --work-dir <dirname> | рабочая директория [по умолчанию: workDir/] |
| -k, --k <int> | длина k-мера (в нуклеотидах, максимум 31) [обязательно] |
| -i, --reads <filenames> | Список файлов с прочтениями. FASTQ, FASTA, gzip- or bzip2-compressed [обязательно] |
| -b, --bad-frequency <int> | максимальная частота встречаемости ошибочных k-меров [по умолчанию: 1] |
Выходные файлы
| файл | описание |
|---|---|
| wd_extract_kmers/kmers/ | папка с k-мерами каждого образца в бинарном формате |
| wd_extract_kmers/stats/ | папка со статистикой |
Преобразует файл таксономической аннотации Kraken2 в CSV формат для совместимости с BandageNG. Может быть визуализирована на графе де Брейна (см. инструкцию).
Входной файл содержит результаты Kraken2 с параметром --use-names. Требуется билиотека ete3.
python3 tax_to_csv.py --class-file=kraken_class.txt --res-file=graph.csvВходные параметры
| параметр | описание |
|---|---|
| --class-file <int> | результат работы Kraken2 |
| --res-file <filename> | имя выходного CSV файла |
Преобразует файл таксономической аннотации MetaPhlAn2 в CSV формат для совместимости с BandageNG. Может быть визуализирована на графе де Брейна (см. инструкцию).
Входной файл содержит промежуточные результаты выравнивания MetaPhlAn2, получаемые например с помощью команды:
metaphlan2.py --input fasta --bowtie2db <path/to/db> -x mpa_v31_CHOCOPhlAn_201901 --nproc 8 --bowtie2out taxa_class.txt wd_unique/contigs_<category>/components.seq.fasta metaphlan2_res.tsvТребуется билиотека ete3.
python3 metaphlan_to_csv.py --class-file=taxa_class.txt --res-file=graph.csvВходные параметры
| параметр | описание |
|---|---|
| --class-file <int> | результат работы MetaPhlAn2 |
| --res-file <filename> | имя выходного CSV файла |
Минимальный пример представлен на странице README.
Полный пример анализа данных представлен в туториале MetaFX.