Home
Welcome to MetaFX wiki page!
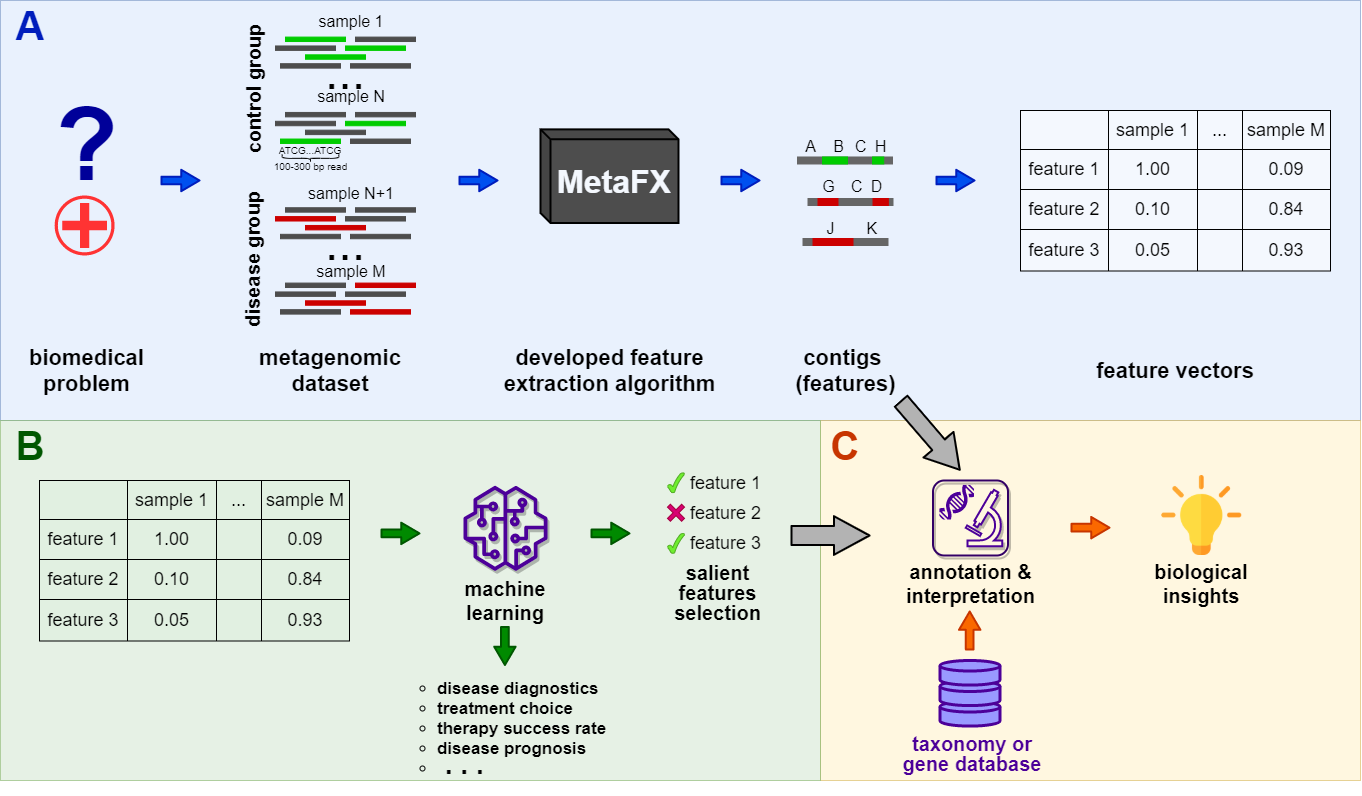
MetaFX (METAgenomic Feature eXtraction) is an open-source library for feature extraction from whole-genome metagenome sequencing data and classification of groups of samples.
The idea behind MetaFX is to introduce the feature extraction algorithm specific for metagenomics short reads data. It is capable of processing hundreds of samples 1-10 Gb each. The distinct property of suggest approach is the construction of meaningful features, which can not only be used to train classification model, but also can be further annotated and biologically interpreted.
To run MetaFX, one need to clone repo with all binaries and add them to PATH.
git clone https://github.com/ivartb/metafx_new
cd metafx_new
export PATH=bin:$PATHRequirements:
- JRE 1.8 or higher
- python3
- python libraries listed in
requirements.txtfile. Can be installed using pip
python -m pip install --upgrade pip
pip install -r requirements.txt- If you want to use
metafx metaspadespipeline, you will also need SPAdes software. Please follow their installation instructions (not recommended for first-time use).
Scripts have been tested under Ubuntu 18.04 LTS and Ubuntu 20.04 LTS, but should generally work on Linux/MacOS.
To run MetaFX use the following syntax:
metafx <pipeline> [<Launch options>] [<Input parameters>]To view the list of supported pipelines run metafx -h or metafx --help.
To view help for launch options and input parameters for selected pipeline run metafx <pipeline> -h or metafx <pipeline> --help.
MetaFX supports both single-end and paired-end input files. For correct detection of paired-end reads, files should be named with suffixes "_R1"&"_R2" or "_r1"&"_r2" after sample name before extension. For example, sample_r1.fastq&sample_r2.fastq, or reads_R1.fq.gz&reads_R2.fq.gz.
By running MetaFX a working directory is created (by default ./workDir/).
All intermediate files and final results are saved there.
MetaFX is a toolbox with a lot of modules divided into three groups:
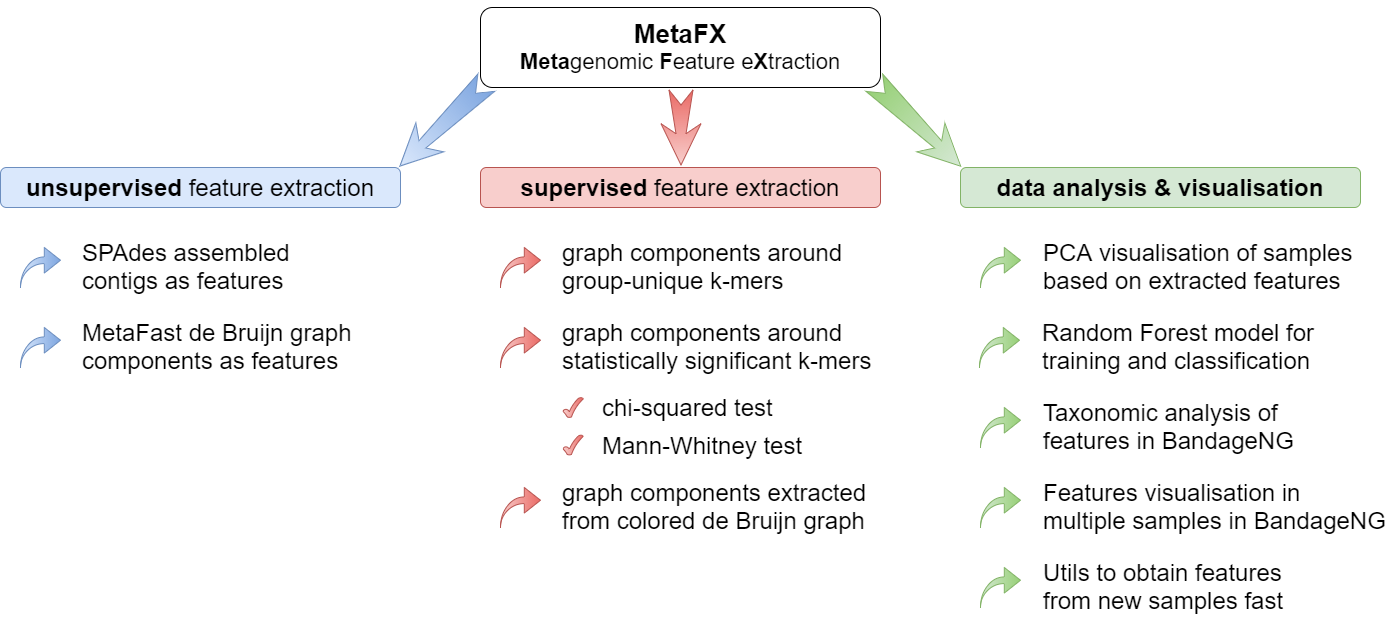
There are pipelines aimed to extract features from metagenomic dataset without any prior knowledge about samples and their relations. Algorithms perform (pseudo-)assembly of samples separately and construct the de Bruijn graph common for all samples. Further, graph components are extracted as features and feature table is constructed.
Performs unsupervised feature extraction and distance estimation via MetaFast (https://github.com/ctlab/metafast/).
metafx metafast -t 2 -m 2G -w wd_metafast -k 21 -i test_data/test*.fastq.gz -b1 100 -b2 500Input parameters
| parameter | description |
|---|---|
| -t, --threads <int> | number of threads to use [default: all] |
| -m, --memory <MEM> | memory to use (values with suffix: 1500M, 4G, etc.) [default: 90% of free RAM] |
| -w, --work-dir <dirname> | working directory [default: workDir/] |
| -k, --k <int> | k-mer size (in nucleotides, maximum value is 31) [mandatory] |
| -i, --reads <filenames> | list of reads files from single environment. FASTQ, FASTA, gzip- or bzip2-compressed [mandatory] |
| -b, --bad-frequency <int> | maximal frequency for a k-mer to be assumed erroneous [default: 1] |
| -l, --min-seq-len <int> | minimal sequence length to be added to a component (in nucleotides) [default: 100] |
| -b1, --min-comp-size <int> | minimum size of extracted components (features) in k-mers [default: 1000] |
| -b2, --max-comp-size <int> | maximum size of extracted components (features) in k-mers [default: 10000] |
| --kmers-dir <dirname> | directory with pre-computed k-mers for samples in binary format [optional] |
Output files
For backward compatibility with supervised feature extraction, every sample belongs to category all.
| file | description |
|---|---|
| wd_metafast/categories_samples.tsv | tab-separated file with 3 columns: <category>\t<present_samples>\t<absent_samples> |
| wd_metafast/samples_categories.tsv | tab-separated file with 2 columns: <sample_name>\t<category> |
| wd_metafast/feature_table.tsv | tab-separated numeric features file: rows – features, columns – samples |
| wd_metafast/contigs_all/seq-builder-many/sequences/component.seq.fasta | contigs in FASTA format as features (suitable for annotation and biological interpretation) |
| wd_metafast/matrices/dist_matrix_<DATE_TIME>.txt | Distance matrix between every pair of samples built over feature table using Bray-Curtis dissimilarity |
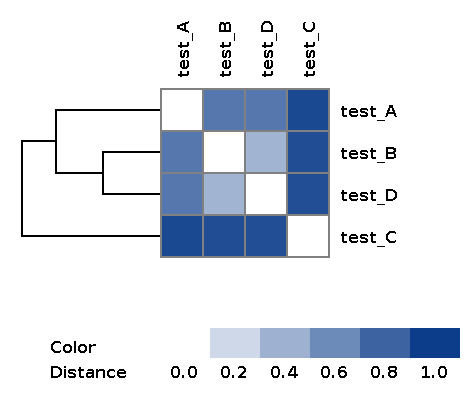
| wd_metafast/matrices/dist_matrix_<DATE_TIME>_heatmap.png | Heatmap and dendrogram of samples based on distance matrix |
Example of the resulting heatmap with dendrogram:
Performs unsupervised feature extraction and distance estimation via metaSpades (https://cab.spbu.ru/software/meta-spades/)
metafx metaspades -t 2 -m 4G -w wd_metaspades -k 21 -i test_data/test_*.fastq.gz -b1 500 -b2 5000Input parameters
| parameter | description |
|---|---|
| -t, --threads <int> | number of threads to use [default: all] |
| -m, --memory <MEM> | memory to use (values with suffix: 1500M, 4G, etc.) [default: 90% of free RAM] |
| -w, --work-dir <dirname> | working directory [default: workDir/] |
| -k, --k <int> | k-mer size (in nucleotides, maximum value is 31) [mandatory] |
| -i, --reads <filenames> | list of PAIRED-END reads files from single environment. FASTQ, FASTA, gzip-compressed [mandatory] |
| --separate | if TRUE use every spades contig as a separate feature (-l, -b1, -b2 ignored) [default: False] |
| -l, --min-seq-len <int> | minimal sequence length to be added to a component (in nucleotides) [default: 100] |
| -b1, --min-comp-size <int> | minimum size of extracted components (features) in k-mers [default: 1000] |
| -b2, --max-comp-size <int> | maximum size of extracted components (features) in k-mers [default: 10000] |
| --kmers-dir <dirname> | directory with pre-computed k-mers for samples in binary format [optional] |
Output files
For backward compatibility with supervised feature extraction, every sample belongs to category all.
| file | description |
|---|---|
| wd_metaspades/categories_samples.tsv | tab-separated file with 3 columns: <category>\t<present_samples>\t<absent_samples> |
| wd_metaspades/samples_categories.tsv | tab-separated file with 2 columns: <sample_name>\t<category> |
| wd_metaspades/feature_table.tsv | tab-separated numeric features file: rows – features, columns – samples |
| wd_metaspades/contigs_all/seq-builder-many/sequences/component.seq.fasta | contigs in FASTA format as features (suitable for annotation and biological interpretation) |
| wd_metaspades/matrices/dist_matrix_<DATE_TIME>*.txt | Distance matrix between every pair of samples built over feature table using Bray-Curtis dissimilarity |
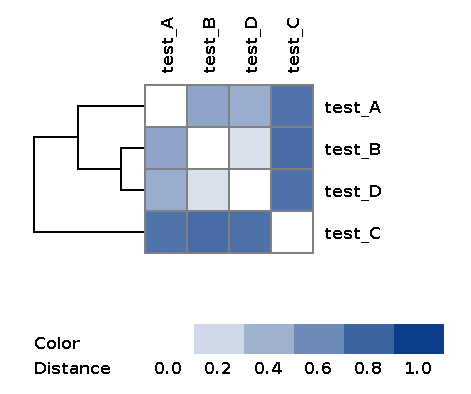
| wd_metaspades/matrices/dist_matrix_<DATE_TIME>*_heatmap.png | Heatmap and dendrogram of samples based on distance matrix |
Example of the resulting heatmap with dendrogram:
There are pipelines aimed to extract group-relevant features based on metadata about samples such as diagnosis, treatment, biochemical results, etc. Dataset is split into groups of samples based on provided metadata information and group-specific features are constructed based on de Bruijn graphs. The resulting features are combined into feature table.
Performs supervised feature extraction using group-specific k-mers. K-mer is considered to be group-specific if it is present in at least G samples of certain group and absent in all other samples.
metafx unique -t 2 -m 4G -w wd_unique -k 21 -i test_data/sample_list_train.txtInput parameters
| parameter | description |
|---|---|
| -t, --threads <int> | number of threads to use [default: all] |
| -m, --memory <MEM> | memory to use (values with suffix: 1500M, 4G, etc.) [default: 90% of free RAM] |
| -w, --work-dir <dirname> | working directory [default: workDir/] |
| -k, --k <int> | k-mer size (in nucleotides, maximum value is 31) [mandatory] |
| -i, --reads <filename> | tab-separated file with 2 values in each row: <path_to_file>\t [mandatory] |
| -b, --bad-frequency <int> | maximal frequency for a k-mer to be assumed erroneous [default: 1] |
| --min-samples <int> | k-mer is considered group-specific if present in at least G samples of that group. G iterates in range [--min-samples; --max-samples] [default: 2] |
| --max-samples <int> | k-mer is considered group-specific if present in at least G samples of that group. G iterates in range [--min-samples; --max-samples] [default: #{samples in category}/2 + 1] |
| --kmers-dir <dirname> | directory with pre-computed k-mers for samples in binary format [optional] |
Output files
| file | description |
|---|---|
| wd_unique/categories_samples.tsv | tab-separated file with 3 columns: <category>\t<present_samples>\t<absent_samples> |
| wd_unique/samples_categories.tsv | tab-separated file with 2 columns: <sample_name>\t<category> |
| wd_unique/feature_table.tsv | tab-separated numeric features file: rows – features, columns – samples |
| wd_unique/contigs_<category>/seq-builder-many/sequences/component.seq.fasta | contigs in FASTA format as features (suitable for annotation and biological interpretation) |
Performs supervised feature extraction using statistically significant k-mers. K-mer significance for a certain group is determined in two steps. Firstly, Pearson's chi-squared test for homogeneity filters out k-mers, which have the same distribution between categories. Secondly, for each pair of categories Mann–Whitney U test is applied to select k-mers with different occurrences between categories.
metafx stats -t 2 -m 4G -w wd_stats -k 21 -i test_data/sample_list.txt --pchi2 0.05 --pmw 0.05Input parameters
| parameter | description |
|---|---|
| -t, --threads <int> | number of threads to use [default: all] |
| -m, --memory <MEM> | memory to use (values with suffix: 1500M, 4G, etc.) [default: 90% of free RAM] |
| -w, --work-dir <dirname> | working directory [default: workDir/] |
| -k, --k <int> | k-mer size (in nucleotides, maximum value is 31) [mandatory] |
| -i, --reads <filename> | tab-separated file with 2 values in each row: <path_to_file>\t [mandatory] |
| -b, --bad-frequency <int> | maximal frequency for a k-mer to be assumed erroneous [default: 1] |
| --phi2 <float> | p-value for chi-squared test [default: 0.05] |
| --pmw <float> | p-value for Mann–Whitney test [default: 0.05] |
| --kmers-dir <dirname> | directory with pre-computed k-mers for samples in binary format [optional] |
Output files
| file | description |
|---|---|
| wd_stats/categories_samples.tsv | tab-separated file with 3 columns: <category>\t<present_samples>\t<absent_samples> |
| wd_stats/samples_categories.tsv | tab-separated file with 2 columns: <sample_name>\t<category> |
| wd_stats/feature_table.tsv | tab-separated numeric features file: rows – features, columns – samples |
| wd_stats/contigs_<category>/seq-builder-many/sequences/component.seq.fasta | contigs in FASTA format as features (suitable for annotation and biological interpretation) |
Performs supervised feature extraction using group-colored de Bruijn graph. All k-mers are colored based on their relative occurrences in categories and further de Bruijn graph is split into colored components.
Important! This module supports up to 3 categories of samples. If you have more, consider using other MetaFX modules.
metafx colored -t 2 -m 4G -w wd_colored -k 21 -i test_data/sample_list_3.txt --linear --n-comps 100 --perc 0.8Input parameters
| parameter | description |
|---|---|
| -t, --threads <int> | number of threads to use [default: all] |
| -m, --memory <MEM> | memory to use (values with suffix: 1500M, 4G, etc.) [default: 90% of free RAM] |
| -w, --work-dir <dirname> | working directory [default: workDir/] |
| -k, --k <int> | k-mer size (in nucleotides, maximum value is 31) [mandatory] |
| -i, --reads <filename> | tab-separated file with 2 values in each row: <path_to_file>\t [mandatory] |
| -b, --bad-frequency <int> | maximal frequency for a k-mer to be assumed erroneous [default: 1] |
| --total-coverage | if TRUE count k-mers occurrences in colored graph as total coverage in samples, otherwise as number of samples [default: False] |
| --separate | if TRUE use only color-specific k-mers in components (does not work in --linear mode) [default: False] |
| --linear | if TRUE extract only linear components choosing the best path on each graph fork [default: False] |
| --n-comps <int> | select not more than components for each category [default: -1, means all components] |
| --perc <float> | relative abundance of k-mer in category to be considered color-specific [default: 0.9] |
| --kmers-dir <dirname> | directory with pre-computed k-mers for samples in binary format [optional] |
Output files
| file | description |
|---|---|
| wd_colored/categories_samples.tsv | tab-separated file with 3 columns: <category>\t<present_samples>\t<absent_samples> |
| wd_colored/samples_categories.tsv | tab-separated file with 2 columns: <sample_name>\t<category> |
| wd_colored/feature_table.tsv | tab-separated numeric features file: rows – features, columns – samples |
| wd_colored/contigs_<category>/seq-builder-many/sequences/component.seq.fasta | contigs in FASTA format as features (suitable for annotation and biological interpretation) |
There are pipelines for analysis of the feature extraction results. Methods for samples similarity visualisation and training machine learning models are implemented. Classification models can be trained to predict samples' properties based on extracted features and to efficiently process new samples from the same environment.
TODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODO
Input parameters
| parameter | description |
|---|---|
Output files
| file | description |
|---|---|
TODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODO
Input parameters
| parameter | description |
|---|---|
Output files
| file | description |
|---|---|
TODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODO
Input parameters
| parameter | description |
|---|---|
Output files
| file | description |
|---|---|
TODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODO
Input parameters
| parameter | description |
|---|---|
Output files
| file | description |
|---|---|
TODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODO
Input parameters
| parameter | description |
|---|---|
Output files
| file | description |
|---|---|
TODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODOTODO
Input parameters
| parameter | description |
|---|---|
Output files
| file | description |
|---|---|
Minimal example is provided in the README page.
More sophisticated example is presented as a jupyter notebook with links to input and output data, all commands and commentaries.