This tutorial introduces Sunny through its features for performing conventional spin wave theory calculations. We consider the crystal CoRh₂O₄ and reproduce the calculations of Ge et al., Phys. Rev. B 96, 064413.
Sunny is implemented in Julia, which allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users should begin with our Getting Started guide. Sunny requires Julia 1.10 or later.
From the Julia prompt, load Sunny and also GLMakie for graphics.
using Sunny, GLMakie
If these packages are not yet installed, Julia will offer to install them. If executing this tutorial gives an error, you may need to update Sunny and GLMakie from the built-in package manager.
The Units object selects reference energy and length scales, and uses these to provide physical constants. For example, units.K returns one kelvin as 0.086 meV, where the Boltzmann constant is implicit.
A crystallographic cell may be loaded from a .cif file, or specified from atom positions and types.
Start by defining the shape of the conventional chemical cell. CoRh₂O₄ has cubic spacegroup 227 (Fd-3m). Its lattice constants are 8.5 Å, and the cell angles are 90°. With this information, lattice_vectors constructs a 3×3 matrix latvecs. Columns of latvecs define the lattice vectors $(𝐚_1, 𝐚_2, 𝐚_3)$ in the global Cartesian coordinate system. Conversely, columns of inv(latvecs) define the global Cartesian axes $(\hat{x}, \hat{y}, \hat{z})$ in components of the lattice vectors.
a = 8.5031 # (Å)
+1. Spin wave simulations of CoRh₂O₄ · Sunny documentation
This tutorial introduces Sunny through its features for performing conventional spin wave theory calculations. We consider the crystal CoRh₂O₄ and reproduce the calculations of Ge et al., Phys. Rev. B 96, 064413.
Sunny is implemented in Julia, which allows for interactive development (like Python or Matlab) while also providing high numerical efficiency (like C++ or Fortran). New Julia users should begin with our Getting Started guide. Sunny requires Julia 1.10 or later.
From the Julia prompt, load Sunny and also GLMakie for graphics.
using Sunny, GLMakie
If these packages are not yet installed, Julia will offer to install them. If executing this tutorial gives an error, you may need to update Sunny and GLMakie from the built-in package manager.
The Units object selects reference energy and length scales, and uses these to provide physical constants. For example, units.K returns one kelvin as 0.086 meV, where the Boltzmann constant is implicit.
A crystallographic cell may be loaded from a .cif file, or specified from atom positions and types.
Start by defining the shape of the conventional chemical cell. CoRh₂O₄ has cubic spacegroup 227 (Fd-3m). Its lattice constants are 8.5 Å, and the cell angles are 90°. With this information, lattice_vectors constructs a 3×3 matrix latvecs. Columns of latvecs define the lattice vectors $(𝐚_1, 𝐚_2, 𝐚_3)$ in the global Cartesian coordinate system. Conversely, columns of inv(latvecs) define the global Cartesian axes $(\hat{x}, \hat{y}, \hat{z})$ in components of the lattice vectors.
a = 8.5031 # (Å)
latvecs = lattice_vectors(a, a, a, 90, 90, 90)
To search for the ground state, call randomize_spins! and minimize_energy! in sequence. For this problem, optimization converges rapidly to the expected Néel order. See this with plot_spins, where spins are colored according to their global $z$-component.
randomize_spins!(sys)
minimize_energy!(sys)
-plot_spins(sys; color=[S[3] for S in sys.dipoles])
The diamond lattice is bipartite, allowing each spin to perfectly anti-align with its 4 nearest-neighbors. Each of these 4 bonds contribute $-J s^2$ to the total energy. Two sites participate in each bond, so the energy per site is $-2 J s^2$. Check this by calling energy_per_site.
The most compact magnetic cell for this Néel order is a primitive unit cell. Reduce the magnetic cell size using reshape_supercell, where columns of the shape matrix are primitive lattice vectors as multiples of the conventional cubic lattice vectors $(𝐚_1, 𝐚_2, 𝐚_3)$. One could alternatively use shape = cryst.latvecs \ cryst.prim_latvecs. Verify that the energy per site is unchanged after the reshaping the supercell.
shape = [0 1 1;
+plot_spins(sys; color=[S[3] for S in sys.dipoles])
The diamond lattice is bipartite, allowing each spin to perfectly anti-align with its 4 nearest-neighbors. Each of these 4 bonds contribute $-J s^2$ to the total energy. Two sites participate in each bond, so the energy per site is $-2 J s^2$. Check this by calling energy_per_site.
The most compact magnetic cell for this Néel order is a primitive unit cell. Reduce the magnetic cell size using reshape_supercell, where columns of the shape matrix are primitive lattice vectors as multiples of the conventional cubic lattice vectors $(𝐚_1, 𝐚_2, 𝐚_3)$. One could alternatively use shape = cryst.latvecs \ cryst.prim_latvecs. Verify that the energy per site is unchanged after the reshaping the supercell.
With this primitive cell, we will perform a SpinWaveTheory calculation of the structure factor $\mathcal{S}(𝐪,ω)$. The measurement ssf_perp indicates projection of the spin structure factor $\mathcal{S}(𝐪,ω)$ perpendicular to the direction of momentum transfer, as appropriate for unpolarized neutron scattering. The isotropic FormFactor for Co²⁺ dampens intensities at large $𝐪$.
With this primitive cell, we will perform a SpinWaveTheory calculation of the structure factor $\mathcal{S}(𝐪,ω)$. The measurement ssf_perp indicates projection of the spin structure factor $\mathcal{S}(𝐪,ω)$ perpendicular to the direction of momentum transfer, as appropriate for unpolarized neutron scattering. The isotropic FormFactor for Co²⁺ dampens intensities at large $𝐪$.
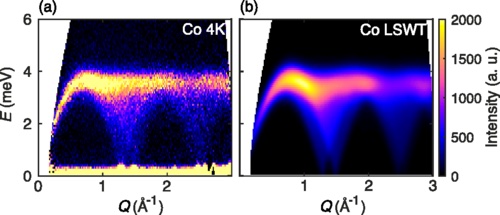
Sometimes experimental data is only available as a powder average, i.e., as an average over all possible crystal orientations. Use powder_average to simulate these intensities. Each $𝐪$-magnitude defines a spherical shell in reciprocal space. Consider 200 radii from 0 to 3 inverse angstroms, and collect 2000 random samples per spherical shell. As configured, this calculation completes in about two seconds. Had we used the conventional cubic cell, the calculation would be an order of magnitude slower.
Sometimes experimental data is only available as a powder average, i.e., as an average over all possible crystal orientations. Use powder_average to simulate these intensities. Each $𝐪$-magnitude defines a spherical shell in reciprocal space. Consider 200 radii from 0 to 3 inverse angstroms, and collect 2000 random samples per spherical shell. As configured, this calculation completes in about two seconds. Had we used the conventional cubic cell, the calculation would be an order of magnitude slower.
radii = range(0, 3, 200) # (1/Å)
res = powder_average(cryst, radii, 2000) do qs
intensities(swt, qs; energies, kernel)
end
-plot_intensities(res; units, saturation=1.0, title="CoRh₂O₄ Powder Average")
This result can be compared to experimental neutron scattering data from Fig. 5 of Ge et al.
Spin wave theory neglects thermal fluctuations of the magnetic order. The next CoRh₂O₄ tutorial demonstrates how to sample spins in thermal equilibrium, and measure dynamical correlations from the classical spin dynamics.
Sunny also offers features that go beyond the dipole approximation of a quantum spin via the theory of SU(N) coherent states. This can be especially useful for systems with strong single-ion anisotropy, as demonstrated in the FeI₂ tutorial.
Settings
This document was generated with Documenter.jl version 1.7.0 on Thursday 19 September 2024. Using Julia version 1.10.5.
Spin wave theory neglects thermal fluctuations of the magnetic order. The next CoRh₂O₄ tutorial demonstrates how to sample spins in thermal equilibrium, and measure dynamical correlations from the classical spin dynamics.
Sunny also offers features that go beyond the dipole approximation of a quantum spin via the theory of SU(N) coherent states. This can be especially useful for systems with strong single-ion anisotropy, as demonstrated in the FeI₂ tutorial.
Settings
This document was generated with Documenter.jl version 1.7.0 on Friday 20 September 2024. Using Julia version 1.10.5.
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-34218f8c.png b/previews/PR318/examples/02_LLD_CoRh2O4-34218f8c.png
deleted file mode 100644
index 8d3c6fdb5..000000000
Binary files a/previews/PR318/examples/02_LLD_CoRh2O4-34218f8c.png and /dev/null differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-389c7e86.png b/previews/PR318/examples/02_LLD_CoRh2O4-389c7e86.png
new file mode 100644
index 000000000..0b5c51ffe
Binary files /dev/null and b/previews/PR318/examples/02_LLD_CoRh2O4-389c7e86.png differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-5a5e47ce.png b/previews/PR318/examples/02_LLD_CoRh2O4-5a5e47ce.png
deleted file mode 100644
index 5e4d8a99d..000000000
Binary files a/previews/PR318/examples/02_LLD_CoRh2O4-5a5e47ce.png and /dev/null differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-66d7beff.png b/previews/PR318/examples/02_LLD_CoRh2O4-66d7beff.png
deleted file mode 100644
index 4a3e433a3..000000000
Binary files a/previews/PR318/examples/02_LLD_CoRh2O4-66d7beff.png and /dev/null differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-687ccd73.png b/previews/PR318/examples/02_LLD_CoRh2O4-687ccd73.png
new file mode 100644
index 000000000..184d17e21
Binary files /dev/null and b/previews/PR318/examples/02_LLD_CoRh2O4-687ccd73.png differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-833db2a2.png b/previews/PR318/examples/02_LLD_CoRh2O4-833db2a2.png
new file mode 100644
index 000000000..d16b79225
Binary files /dev/null and b/previews/PR318/examples/02_LLD_CoRh2O4-833db2a2.png differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-866343c2.png b/previews/PR318/examples/02_LLD_CoRh2O4-866343c2.png
deleted file mode 100644
index c8c4cda42..000000000
Binary files a/previews/PR318/examples/02_LLD_CoRh2O4-866343c2.png and /dev/null differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-867ba07b.png b/previews/PR318/examples/02_LLD_CoRh2O4-867ba07b.png
deleted file mode 100644
index 2fb7c481e..000000000
Binary files a/previews/PR318/examples/02_LLD_CoRh2O4-867ba07b.png and /dev/null differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-8ae4e334.png b/previews/PR318/examples/02_LLD_CoRh2O4-8ae4e334.png
deleted file mode 100644
index 4dcd5feab..000000000
Binary files a/previews/PR318/examples/02_LLD_CoRh2O4-8ae4e334.png and /dev/null differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-8beeef47.png b/previews/PR318/examples/02_LLD_CoRh2O4-8beeef47.png
deleted file mode 100644
index 6faf58a93..000000000
Binary files a/previews/PR318/examples/02_LLD_CoRh2O4-8beeef47.png and /dev/null differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-8c71a528.png b/previews/PR318/examples/02_LLD_CoRh2O4-8c71a528.png
new file mode 100644
index 000000000..6b577e986
Binary files /dev/null and b/previews/PR318/examples/02_LLD_CoRh2O4-8c71a528.png differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-8eca46a8.png b/previews/PR318/examples/02_LLD_CoRh2O4-8eca46a8.png
new file mode 100644
index 000000000..7bcd733c7
Binary files /dev/null and b/previews/PR318/examples/02_LLD_CoRh2O4-8eca46a8.png differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-d608e0c1.png b/previews/PR318/examples/02_LLD_CoRh2O4-d608e0c1.png
new file mode 100644
index 000000000..974a1260e
Binary files /dev/null and b/previews/PR318/examples/02_LLD_CoRh2O4-d608e0c1.png differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4-ebc60cb1.png b/previews/PR318/examples/02_LLD_CoRh2O4-ebc60cb1.png
new file mode 100644
index 000000000..e740bbc61
Binary files /dev/null and b/previews/PR318/examples/02_LLD_CoRh2O4-ebc60cb1.png differ
diff --git a/previews/PR318/examples/02_LLD_CoRh2O4.html b/previews/PR318/examples/02_LLD_CoRh2O4.html
index 0fb438df2..1b1bad9ee 100644
--- a/previews/PR318/examples/02_LLD_CoRh2O4.html
+++ b/previews/PR318/examples/02_LLD_CoRh2O4.html
@@ -11,16 +11,16 @@
set_exchange!(sys, J, Bond(1, 3, [0,0,0]))
randomize_spins!(sys)
minimize_energy!(sys)
-plot_spins(sys; color=[S[3] for S in sys.dipoles])
Use repeat_periodically to extend the system to 10×10×10 chemical unit cells. The ground state Néel order is retained. Increasing the system size further would reduce finite-size artifacts and increase momentum-space resolution, but would also make the simulations slower.
sys = repeat_periodically(sys, (10, 10, 10))
-plot_spins(sys; color=[S[3] for S in sys.dipoles])
We will be using a Langevin spin dynamics to thermalize the system. This dynamics is a variant of the Landau-Lifshitz equation that incorporates noise and dissipation terms, which are linked by a fluctuation-dissipation theorem. The temperature 6 K ≈ 1.38 meV is slightly above ordering for this model. The dimensionless damping magnitude sets a timescale for coupling to the implicit thermal bath; 0.2 is usually a good choice.
langevin = Langevin(; damping=0.2, kT=16*units.K)
Langevin(<missing>; damping=0.2, kT=1.3787733219432283)
+plot_spins(sys; color=[S[3] for S in sys.dipoles])
Use repeat_periodically to extend the system to 10×10×10 chemical unit cells. The ground state Néel order is retained. Increasing the system size further would reduce finite-size artifacts and increase momentum-space resolution, but would also make the simulations slower.
sys = repeat_periodically(sys, (10, 10, 10))
+plot_spins(sys; color=[S[3] for S in sys.dipoles])
We will be using a Langevin spin dynamics to thermalize the system. This dynamics is a variant of the Landau-Lifshitz equation that incorporates noise and dissipation terms, which are linked by a fluctuation-dissipation theorem. The temperature 6 K ≈ 1.38 meV is slightly above ordering for this model. The dimensionless damping magnitude sets a timescale for coupling to the implicit thermal bath; 0.2 is usually a good choice.
Use suggest_timestep to select an integration timestep. A dimensionless error tolerance of 1e-2 is usually a good choice. The suggested timestep will vary according to the magnetic configuration. It is reasonable to start from an energy-minimized configuration.
Consider dt ≈ 0.02528 for this spin configuration at tol = 0.01.
Now run a Langevin trajectory to sample spin configurations. Keep track of the energy per site at each time step.
energies = [energy_per_site(sys)]
for _ in 1:1000
step!(sys, langevin)
push!(energies, energy_per_site(sys))
end
From the relaxed spin configuration, we can learn that dt was a little smaller than necessary; increasing it will make the remaining simulations faster.
Plot the spins colored by their alignment with a reference spin at the origin. The field sys.dipoles is a 4D array storing the spin dipole data. The first three indices of label the chemical cell, while the fourth index labels an atom within the cell. Note that Julia arrays use 1-based indexing. Thermal fluctuations are apparent in the plot.
S0 = sys.dipoles[1,1,1,1]
-plot_spins(sys; color=[S'*S0 for S in sys.dipoles])
Use SampledCorrelationsStatic to estimate spatial correlations for configurations in classical thermal equilibrium. Each call to add_sample! will accumulate data for the current spin snapshot.
Plot the spins colored by their alignment with a reference spin at the origin. The field sys.dipoles is a 4D array storing the spin dipole data. The first three indices of label the chemical cell, while the fourth index labels an atom within the cell. Note that Julia arrays use 1-based indexing. Thermal fluctuations are apparent in the plot.
S0 = sys.dipoles[1,1,1,1]
+plot_spins(sys; color=[S'*S0 for S in sys.dipoles])
Use SampledCorrelationsStatic to estimate spatial correlations for configurations in classical thermal equilibrium. Each call to add_sample! will accumulate data for the current spin snapshot.
Collect 20 additional samples. Perform 100 Langevin time-steps between measurements to approximately decorrelate the sample in thermal equilibrium.
for _ in 1:20
@@ -29,7 +29,7 @@
end
add_sample!(sc, sys)
end
Use q_space_grid to define a slice of momentum space $[H, K, 0]$, where $H$ and $K$ each range from -10 to 10 in RLU. This command produces a 200×200 grid of sample points.
Calculate and plot the instantaneous structure factor on the slice by integrating over all energy values ω. We employ the appropriate FormFactor for Co2⁺. Selecting saturation = 1.0 sets the color saturation point to the maximum intensity value. This is reasonable because we are above the ordering temperature, and do not have sharp Bragg peaks.
res = intensities_static(sc, grid)
-plot_intensities(res; saturation=1.0, title="Static Intensities at T = 16 K")
To collect statistics for the dynamical structure factor intensities $\mathcal{S}(𝐪,ω)$ at finite temperature, use SampledCorrelations. It requires a range of energies to resolve, which will be associated with frequencies of the classical spin dynamics. The integration timestep dt can be somewhat larger than that used by the Langevin dynamics.
dt = 2*langevin.dt
+plot_intensities(res; saturation=1.0, title="Static Intensities at T = 16 K")
To collect statistics for the dynamical structure factor intensities $\mathcal{S}(𝐪,ω)$ at finite temperature, use SampledCorrelations. It requires a range of energies to resolve, which will be associated with frequencies of the classical spin dynamics. The integration timestep dt can be somewhat larger than that used by the Langevin dynamics.
Define spherical shells in reciprocal space via their radii, in absolute units of 1/Å. For each shell, calculate and average the intensities at 350 $𝐪$-points
Define spherical shells in reciprocal space via their radii, in absolute units of 1/Å. For each shell, calculate and average the intensities at 350 $𝐪$-points
radii = range(0, 3.5, 200) # (1/Å)
res = powder_average(cryst, radii, 350) do qs
intensities(sc, qs; energies, langevin.kT)
end
-plot_intensities(res; units, title="Powder Average at 16 K")
Settings
This document was generated with Documenter.jl version 1.7.0 on Thursday 19 September 2024. Using Julia version 1.10.5.