Indexing and Visualization for AMRs and Virulence Genes in Genomes and Metagenomes
Develop a tool capable of evaluating metadata quality in the Sequence Read Archive (SRA), together with the contigs assembled from each of the runs (SRRs).
Provide visualization capabilities to assist in downstream interpretation.
Use cases:
- Find accession/contig by taxonomy
- Find accessions/contig by gene (drug resistance, virulence factor)
- Find accession/contig by genes cluster
- Find accession/contig by domain
- Find accession by host
- Find similar SRR/contigs
- Find accession/contig by sequence
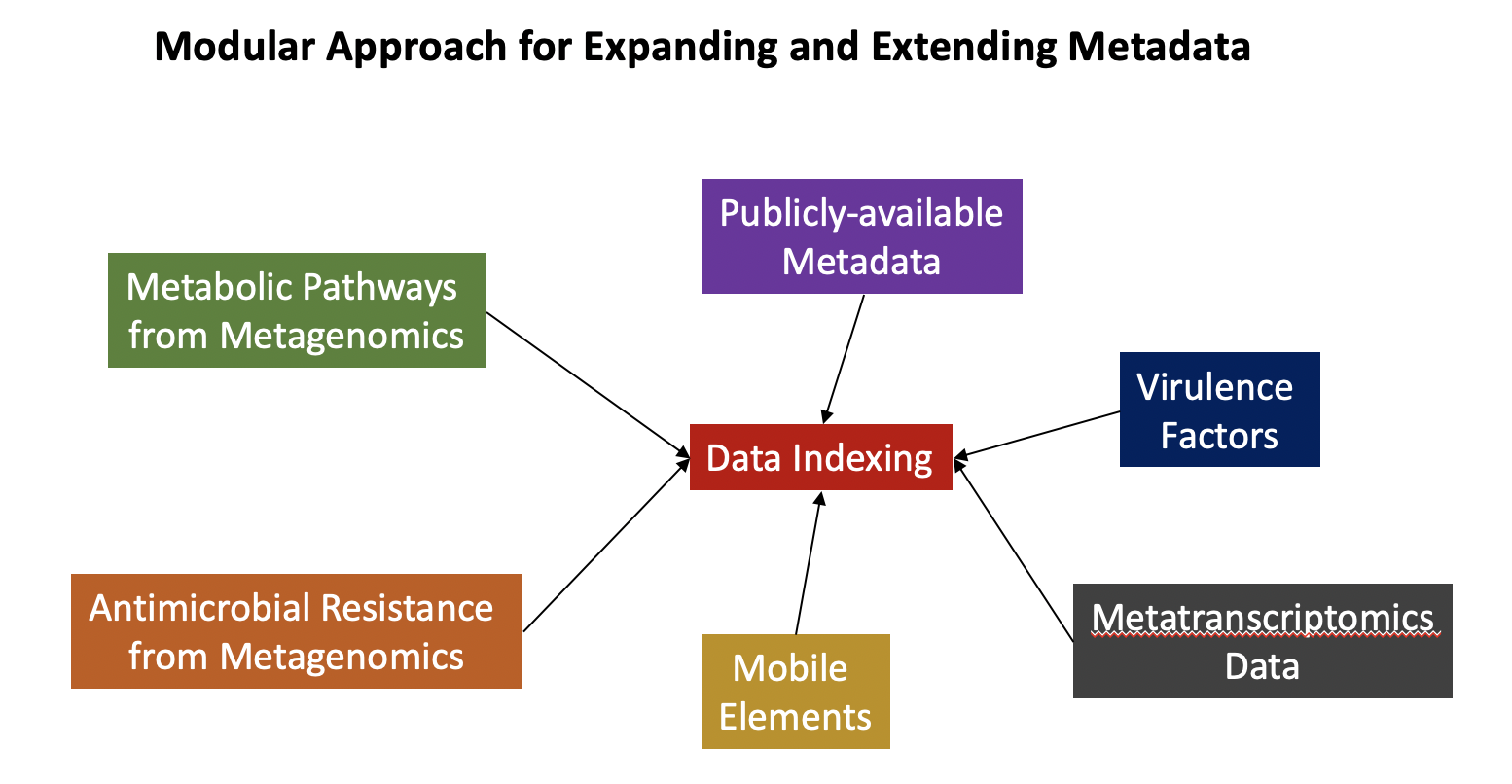
The overarching framework of our approach is to enable the integration of various metadata types in a large data structure that consists of multiple tables. An example of such a framework and the various data types that can be supported can be seen below.
Q: Should similarity/difference in sequence content correlate with similarity/difference in metadata?
A: Let's find out what it looks like currently!
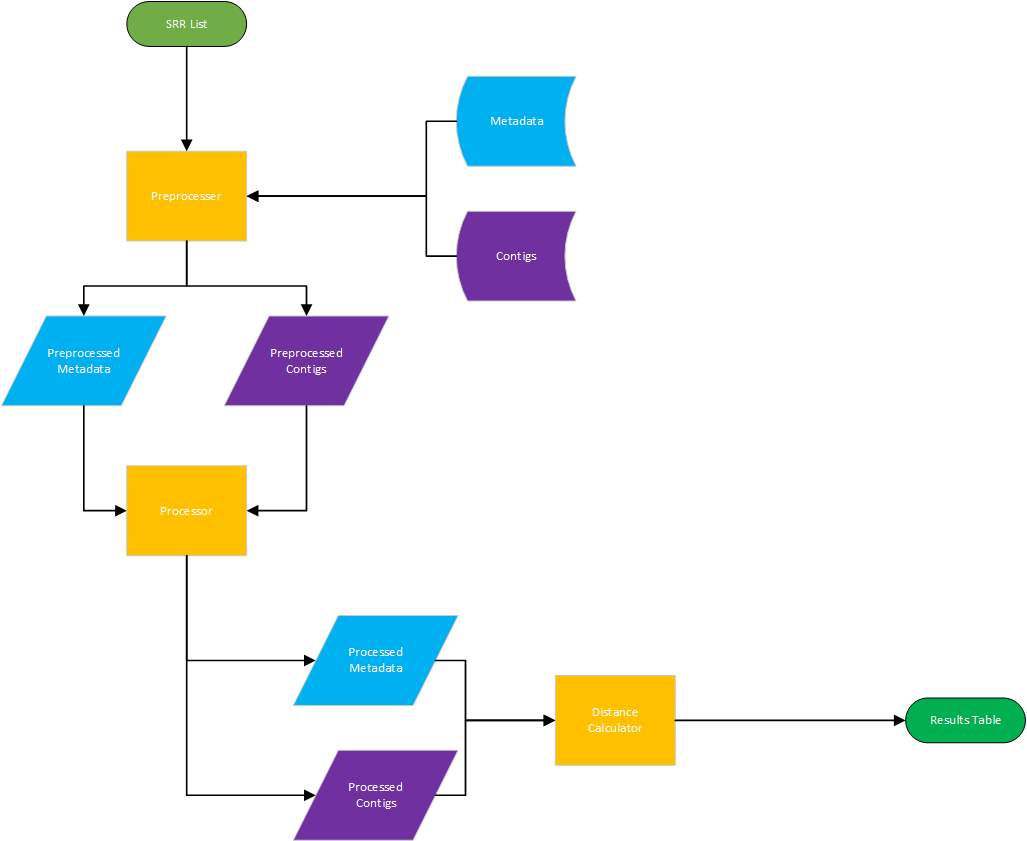
Please see the README files for replicating the analysis described in workflow #2. Note that Python3, R, and access to the Google Cloud Platform will be required.
Eric Holloway, Sergey Nurk, Brett E. Pickett, Ryan Connor